- Calcium & bone metabolism
- Bone Loss after Solid Organ Transplantation: A Review of Organ-Specific Considerations
-
Kyoung Jin Kim, Jeonghoon Ha, Sang Wan Kim, Jung-Eun Kim, Sihoon Lee, Han Seok Choi, Namki Hong, Sung Hye Kong, Seong Hee Ahn, So Young Park, Ki-Hyun Baek, on Behalf of Metabolic Bone Disease Study Group of Korean Endocrine Society
-
Endocrinol Metab. 2024;39(2):267-282. Published online April 25, 2024
-
DOI: https://doi.org/10.3803/EnM.2024.1939
-
-
 Abstract Abstract
 PDF PDF
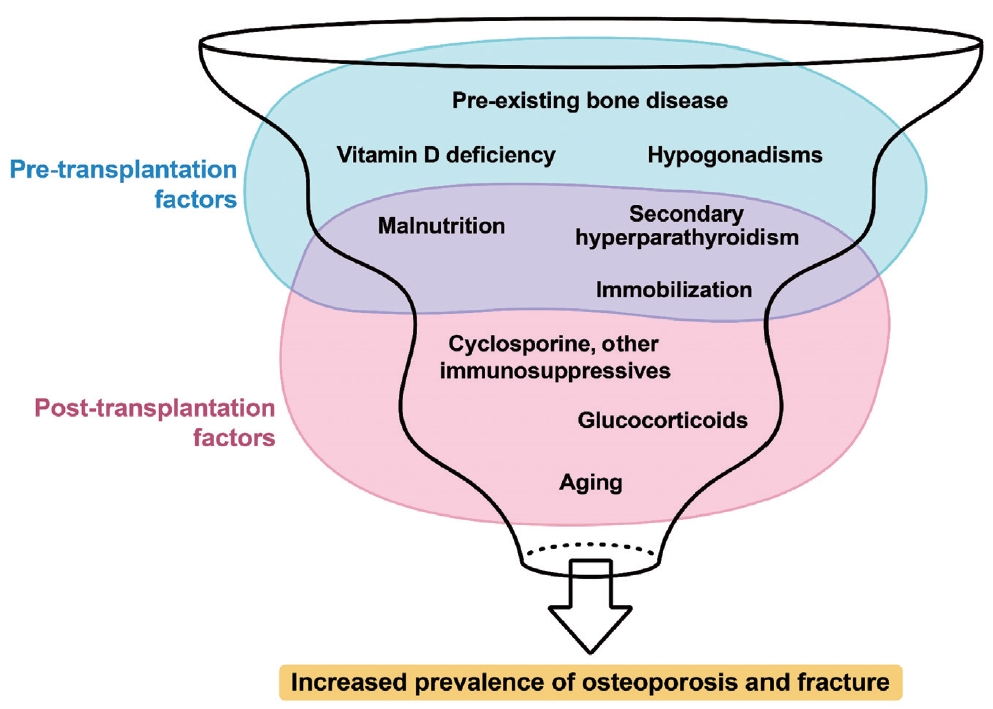
- This review article investigates solid organ transplantation-induced osteoporosis, a critical yet often overlooked issue, emphasizing its significance in post-transplant care. The initial sections provide a comprehensive understanding of the prevalence and multifactorial pathogenesis of transplantation osteoporosis, including factors such as deteriorating post-transplantation health, hormonal changes, and the impact of immunosuppressive medications. Furthermore, the review is dedicated to organ-specific considerations in transplantation osteoporosis, with separate analyses for kidney, liver, heart, and lung transplantations. Each section elucidates the unique challenges and management strategies pertinent to transplantation osteoporosis in relation to each organ type, highlighting the necessity of an organ-specific approach to fully understand the diverse manifestations and implications of transplantation osteoporosis. This review underscores the importance of this topic in transplant medicine, aiming to enhance awareness and knowledge among clinicians and researchers. By comprehensively examining transplantation osteoporosis, this study contributes to the development of improved management and care strategies, ultimately leading to improved patient outcomes in this vulnerable group. This detailed review serves as an essential resource for those involved in the complex multidisciplinary care of transplant recipients.
- Adrenal Gland
- Metabolic Subtyping of Adrenal Tumors: Prospective Multi-Center Cohort Study in Korea
-
Eu Jeong Ku, Chaelin Lee, Jaeyoon Shim, Sihoon Lee, Kyoung-Ah Kim, Sang Wan Kim, Yumie Rhee, Hyo-Jeong Kim, Jung Soo Lim, Choon Hee Chung, Sung Wan Chun, Soon-Jib Yoo, Ohk-Hyun Ryu, Ho Chan Cho, A Ram Hong, Chang Ho Ahn, Jung Hee Kim, Man Ho Choi
-
Endocrinol Metab. 2021;36(5):1131-1141. Published online October 21, 2021
-
DOI: https://doi.org/10.3803/EnM.2021.1149
-
-
5,106
View
-
209
Download
-
8
Web of Science
-
8
Crossref
-
Abstract
PDF
 Supplementary Material Supplementary Material PubReader PubReader  ePub ePub
- Background
Conventional diagnostic approaches for adrenal tumors require multi-step processes, including imaging studies and dynamic hormone tests. Therefore, this study aimed to discriminate adrenal tumors from a single blood sample based on the combination of liquid chromatography-mass spectrometry (LC-MS) and machine learning algorithms in serum profiling of adrenal steroids.
Methods
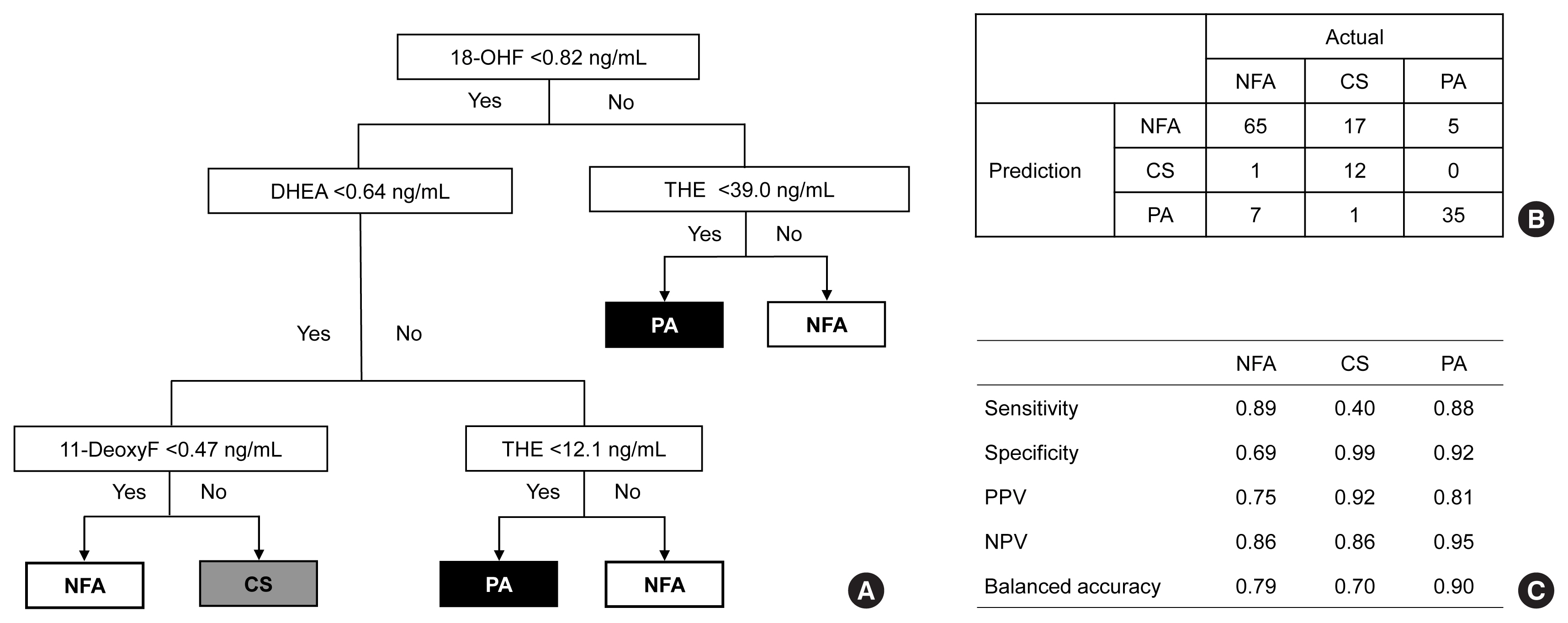
The LC-MS-based steroid profiling was applied to serum samples obtained from patients with nonfunctioning adenoma (NFA, n=73), Cushing’s syndrome (CS, n=30), and primary aldosteronism (PA, n=40) in a prospective multicenter study of adrenal disease. The decision tree (DT), random forest (RF), and extreme gradient boost (XGBoost) were performed to categorize the subtypes of adrenal tumors.
Results
The CS group showed higher serum levels of 11-deoxycortisol than the NFA group, and increased levels of tetrahydrocortisone (THE), 20α-dihydrocortisol, and 6β-hydroxycortisol were found in the PA group. However, the CS group showed lower levels of dehydroepiandrosterone (DHEA) and its sulfate derivative (DHEA-S) than both the NFA and PA groups. Patients with PA expressed higher serum 18-hydroxycortisol and DHEA but lower THE than NFA patients. The balanced accuracies of DT, RF, and XGBoost for classifying each type were 78%, 96%, and 97%, respectively. In receiver operating characteristics (ROC) analysis for CS, XGBoost, and RF showed a significantly greater diagnostic power than the DT. However, in ROC analysis for PA, only RF exhibited better diagnostic performance than DT.
Conclusion
The combination of LC-MS-based steroid profiling with machine learning algorithms could be a promising one-step diagnostic approach for the classification of adrenal tumor subtypes.
-
Citations
Citations to this article as recorded by  - Treating Primary Aldosteronism-Induced Hypertension: Novel Approaches and Future Outlooks
Nathan Mullen, James Curneen, Padraig T Donlon, Punit Prakash, Irina Bancos, Mark Gurnell, Michael C Dennedy
Endocrine Reviews.2024; 45(1): 125. CrossRef - Steroid profiling in adrenal disease
Danni Mu, Dandan Sun, Xia Qian, Xiaoli Ma, Ling Qiu, Xinqi Cheng, Songlin Yu
Clinica Chimica Acta.2024; 553: 117749. CrossRef - Serum and hair steroid profiles in patients with nonfunctioning pituitary adenoma undergoing surgery: A prospective observational study
Seung Shin Park, Yong Hwy Kim, Ho Kang, Chang Ho Ahn, Dong Jun Byun, Man Ho Choi, Jung Hee Kim
The Journal of Steroid Biochemistry and Molecular Biology.2023; 230: 106276. CrossRef - Recent Updates on the Management of Adrenal Incidentalomas
Seung Shin Park, Jung Hee Kim
Endocrinology and Metabolism.2023; 38(4): 373. CrossRef - LC-MS based simultaneous profiling of adrenal hormones of steroids, catecholamines, and metanephrines
Jongsung Noh, Chaelin Lee, Jung Hee Kim, Seung Woon Myung, Man Ho Choi
Journal of Lipid Research.2023; 64(11): 100453. CrossRef - 2023 Korean Endocrine Society Consensus Guidelines for the Diagnosis and Management of Primary Aldosteronism
Jeonghoon Ha, Jung Hwan Park, Kyoung Jin Kim, Jung Hee Kim, Kyong Yeun Jung, Jeongmin Lee, Jong Han Choi, Seung Hun Lee, Namki Hong, Jung Soo Lim, Byung Kwan Park, Jung-Han Kim, Kyeong Cheon Jung, Jooyoung Cho, Mi-kyung Kim, Choon Hee Chung
Endocrinology and Metabolism.2023; 38(6): 597. CrossRef - Toward Systems-Level Metabolic Analysis in Endocrine Disorders and Cancer
Aliya Lakhani, Da Hyun Kang, Yea Eun Kang, Junyoung O. Park
Endocrinology and Metabolism.2023; 38(6): 619. CrossRef - Prevalence and Characteristics of Adrenal Tumors in an Unselected Screening Population
Ying Jing, Jinbo Hu, Rong Luo, Yun Mao, Zhixiao Luo, Mingjun Zhang, Jun Yang, Ying Song, Zhengping Feng, Zhihong Wang, Qingfeng Cheng, Linqiang Ma, Yi Yang, Li Zhong, Zhipeng Du, Yue Wang, Ting Luo, Wenwen He, Yue Sun, Fajin Lv, Qifu Li, Shumin Yang
Annals of Internal Medicine.2022; 175(10): 1383. CrossRef
- Miscellaneous
- Rare PTH Gene Mutations Causing Parathyroid Disorders: A Review
-
Joon-Hyop Lee, Munkhtugs Davaatseren, Sihoon Lee
-
Endocrinol Metab. 2020;35(1):64-70. Published online March 19, 2020
-
DOI: https://doi.org/10.3803/EnM.2020.35.1.64
-
-
5,088
View
-
107
Download
-
8
Web of Science
-
7
Crossref
-
Abstract
PDFPubReader ePub
Since parathyroid hormone (PTH) was first isolated and its gene (PTH) was sequenced, only eight PTH mutations have been discovered. The C18R mutation in PTH, discovered in 1990, was the first to be reported. This autosomal dominant mutation induces endoplasmic reticulum stress and subsequent apoptosis in parathyroid cells. The next mutation, which was reported in 1992, is associated with exon skipping. The substitution of G with C in the first nucleotide of the second intron results in the exclusion of the second exon; since this exon includes the initiation codon, translation initiation is prevented. An S23P mutation and an S23X mutation at the same residue were reported in 1999 and 2012, respectively. Both mutations resulted in hypoparathyroidism. In 2008, a somatic R83X mutation was detected in a parathyroid adenoma tissue sample collected from a patient with hyperparathyroidism. In 2013, a heterozygous p.Met1_Asp6del mutation was incidentally discovered in a case-control study. Two years later, the R56C mutation was reported; this is the only reported hypoparathyroidism-causing mutation in the mature bioactive part of PTH. In 2017, another heterozygous mutation, M14K, was detected. The discovery of these eight mutations in the PTH gene has provided insights into its function and broadened our understanding of the molecular mechanisms underlying mutation progression. Further attempts to detect other such mutations will help elucidate the functions of PTH in a more sophisticated manner. -
Citations
Citations to this article as recorded by - The Intricacies of Renal Phosphate Reabsorption—An Overview
Valerie Walker
International Journal of Molecular Sciences.2024; 25(9): 4684. CrossRef - Molecular and Clinical Spectrum of Primary Hyperparathyroidism
Smita Jha, William F Simonds
Endocrine Reviews.2023; 44(5): 779. CrossRef - Rare cause of persistent hypocalcaemia in infancy due to PTH gene mutation
Savita Khadse, Vrushali Satish Takalikar, Radha Ghildiyal, Nikhil Shah
BMJ Case Reports.2023; 16(9): e256358. CrossRef - Homozygous Ser-1 to Pro-1 mutation in parathyroid hormone identified in hypocalcemic patients results in secretion of a biologically inactive pro-hormone
Patrick Hanna, Ashok Khatri, Shawn Choi, Severine Brabant, Matti L. Gild, Marie L. Piketty, Bruno Francou, Dominique Prié, John T. Potts, Roderick J. Clifton-Bligh, Agnès Linglart, Thomas J. Gardella, Harald Jüppner
Proceedings of the National Academy of Sciences.2023;[Epub] CrossRef - Genetics of monogenic disorders of calcium and bone metabolism
Paul J. Newey, Fadil M. Hannan, Abbie Wilson, Rajesh V. Thakker
Clinical Endocrinology.2022; 97(4): 483. CrossRef - Homozygous missense variant of PTH (c.166C>T, p.(Arg56Cys)) as the cause of familial isolated hypoparathyroidism in a three-year-old child
Stine Linding Andersen, Anja Lisbeth Frederiksen, Astrid Bruun Rasmussen, Mette Madsen, Ann-Margrethe Rønholt Christensen
Journal of Pediatric Endocrinology and Metabolism.2022; 35(5): 691. CrossRef - Novel PTH Gene Mutations Causing Isolated Hypoparathyroidism
Colin P Hawkes, Jamal M Al Jubeh, Dong Li, Susan E Tucker, Tara Rajiyah, Michael A Levine
The Journal of Clinical Endocrinology & Metabolism.2022; 107(6): e2449. CrossRef
- Miscellaneous
- Novel Mutation in PTHLH Related to Brachydactyly Type E2 Initially Confused with Unclassical Pseudopseudohypoparathyroidism
-
Jihong Bae, Hong Seok Choi, So Young Park, Do-Eun Lee, Sihoon Lee
-
Endocrinol Metab. 2018;33(2):252-259. Published online June 21, 2018
-
DOI: https://doi.org/10.3803/EnM.2018.33.2.252
-
-
4,428
View
-
65
Download
-
10
Web of Science
-
9
Crossref
-
Abstract
PDFPubReader ePub
- Background
Autosomal-dominant brachydactyly type E is a congenital abnormality characterized by small hands and feet, which is a consequence of shortened metacarpals and metatarsals. We recently encountered a young gentleman exhibiting shortening of 4th and 5th fingers and toes. Initially, we suspected him having pseudopseudohypoparathyroidism (PPHP) because of normal biochemical parameters, including electrolyte, Ca, P, and parathyroid hormone (PTH) levels; however, his mother and maternal grandmother had the same conditions in their hands and feet. Furthermore, his mother showed normal biochemical parameters. To the best of our knowledge, PPHP is inherited via a mutated paternal allele, owing to the paternal imprinting of GNAS (guanine nucleotide binding protein, alpha stimulating) in the renal proximal tubule. Therefore, we decided to further analyze the genetic background in this family. MethodsWhole exome sequencing was performed using genomic DNA from the affected mother, son, and the unaffected father as a negative control. ResultsWe selected the intersection between 45,490 variants from the mother and 45,646 variants from the son and excluded 27,512 overlapping variants identified from the father. By excluding homogenous and compound heterozygous variants and removing all previously reported variants, 147 variants were identified to be shared by the mother and son. Variants that had least proximities among species were excluded and finally 23 variants remained. ConclusionAmong them, we identified a defect in parathyroid hormone like hormone (PTHLH), encoding the PTH-related protein, to be disease-causative. Herein, we report a family affected with brachydactyly type E2 caused by a novel PTHLH mutation, which was confused with PPHP with unclassical genetic penetrance.
-
Citations
Citations to this article as recorded by - A novel heterozygous mutation in PTHLH causing autosomal dominant brachydactyly type E complicated with short stature
Jian Sun, Nian Yang, Zhengquan Xu, Hongbo Cheng, Xiangxin Zhang
Molecular Genetics & Genomic Medicine.2024;[Epub] CrossRef - A novel mutation in PTHLH in a family with a variable phenotype with brachydactyly, short stature, oligodontia and developmental delay
Mirjam E.A. Scheffer-Rath, Hermine E. Veenstra-Knol, Annemieke M. Boot
Bone Reports.2023; 19: 101699. CrossRef - Bioactive phytoconstituents as potent inhibitors of casein kinase-2: dual implications in cancer and COVID-19 therapeutics
Farah Anjum, Md Nayab Sulaimani, Alaa Shafie, Taj Mohammad, Ghulam Md. Ashraf, Anwar L. Bilgrami, Fahad A. Alhumaydhi, Suliman A. Alsagaby, Dharmendra Kumar Yadav, Md. Imtaiyaz Hassan
RSC Advances.2022; 12(13): 7872. CrossRef - Characterization and expression profiling of G protein-coupled receptors (GPCRs) in Spodoptera litura (Lepidoptera: Noctuidae)
Yanxiao Li, Han Gao, Hui Zhang, Runnan Yu, Fan Feng, Jing Tang, Bin Li
Comparative Biochemistry and Physiology Part D: Genomics and Proteomics.2022; 44: 101018. CrossRef - Genes with specificity for expression in the round cell layer of the growth plate are enriched in genomewide association study (GWAS) of human height
Nora E. Renthal, Priyanka Nakka, John M. Baronas, Henry M. Kronenberg, Joel N. Hirschhorn
Journal of Bone and Mineral Research.2020; 36(12): 2300. CrossRef - Search for Novel Mutational Targets in Human Endocrine Diseases
So Young Park, Myeong Han Seo, Sihoon Lee
Endocrinology and Metabolism.2019; 34(1): 23. CrossRef - A Heterozygous Splice-Site Mutation in PTHLH Causes Autosomal Dominant Shortening of Metacarpals and Metatarsals
Monica Reyes, Bert Bravenboer, Harald Jüppner
Journal of Bone and Mineral Research.2019; 34(3): 482. CrossRef - A 3.06-Mb interstitial deletion on 12p11.22-12.1 caused brachydactyly type E combined with pectus carinatum
Jia Huang, Hong-Yan Liu, Rong-Rong Wang, Hai Xiao, Dong Wu, Tao Li, Ying-Hai Jiang, Xue Zhang
Chinese Medical Journal.2019; 132(14): 1681. CrossRef - Parathyroid Hormone-Related Protein in the Hand or Out of Hand?
Sang Wan Kim
Endocrinology and Metabolism.2018; 33(2): 202. CrossRef
- Adrenal gland
- Genetic Analysis of Multiple Endocrine Neoplasia Type 1 (MEN1) Leads to Misdiagnosis of an Extremely Rare Presentation of Intrasellar Cavernous Hemangioma as MEN1
-
Dong Min Lee, Seung Hee Yu, Hyun Hwa Yoon, Kang Lock Lee, Young Sil Eom, Kiyoung Lee, Byung-Joon Kim, Yeun Sun Kim, Ie Byung Park, Kwang-Won Kim, Sihoon Lee
-
Endocrinol Metab. 2014;29(2):146-153. Published online June 26, 2014
-
DOI: https://doi.org/10.3803/EnM.2014.29.2.146
-
-
3,773
View
-
37
Download
-
1
Web of Science
-
2
Crossref
-
Abstract
PDFPubReader
- Background
Multiple endocrine neoplasia type 1 (MEN1) is a rare inherited disorder characterized by the simultaneous occurrence of endocrine tumors in target tissues (mainly the pituitary, endocrine pancreas, and parathyroid glands). MEN1 is caused by mutations in the MEN1 gene, which functions as a tumor suppressor and consists of one untranslated exon and nine exons encoding the menin protein. This condition is usually suspected when we encounter patients diagnosed with tumors in multiple endocrine organs, as mentioned above. MethodsA 65-year-old woman who underwent surgery for a pancreatic tumor (serous cystadenoma) 5 years previously was referred to our hospital due to neurologic symptoms of diplopia and left ptosis. Brain magnetic resonance imaging revealed a 3.4-cm lesion originating from the cavernous sinus wall and extending into the sellar region. It was thought to be a nonfunctioning tumor from the results of the combined pituitary function test. Incidentally, we found that she also had a pancreatic tumor, indicating the necessity of genetic analysis for MEN1. ResultsGenomic analysis using peripheral leukocytes revealed a heterozygous c.1621G>A mutation in the MEN1 gene that was previously reported to be either a pathogenic mutation or a simple polymorphism. We pursued a stereotactic approach to the pituitary lesion, and microscopic findings of the tumor revealed it to be an intrasellar cavernous hemangioma, a rare finding in the sellar region and even rarer in relation to oculomotor palsy. The patient recovered well from surgery, but refused further evaluation for the pancreatic lesion. ConclusionThere is great emphasis placed on genetic testing in the diagnosis of MEN1, but herein we report a case where it did not assist in diagnosis, hence, further discussion on the role of genetic testing in this disease is needed. Also, in cases of pituitary tumor with cranial nerve palsy, despite its low prevalence, intrasellar cavernous hemangioma could be suspected.
-
Citations
Citations to this article as recorded by - Diffuse cavernous hemangioma of the skull misdiagnosed as skull metastasis in breast cancer patient: one case report and literature review
Huizhi Liu, Xiaojing Chang, Hua Shang, Feng Li, Huandi Zhou, Xiaoying Xue
BMC Cancer.2019;[Epub] CrossRef - Articles in 'Endocrinology and Metabolism' in 2014
Won-Young Lee
Endocrinology and Metabolism.2015; 30(1): 47. CrossRef
- A Case of the Type B Insulin Resistance Syndrome with Chronic Hepatitis B.
-
Hyun Seok Choi, Byoung Ho Choi, Seok Hoo Jeong, Shung Han Choi, Dong Su Shin, Sei hyun Kim, Young Sil Eom, Sihoon Lee, Yeun Sun Kim, Ie Byung Park, Ki Young Lee
-
Endocrinol Metab. 2011;26(4):360-363. Published online December 1, 2011
-
DOI: https://doi.org/10.3803/EnM.2011.26.4.360
-
-
Abstract
PDF
- Type B insulin resistance syndrome is rare autoimmune disease that is characterized by various abnormalities of glycemic homeostasis, from hyperglycemia caused by extreme insulin resistance to fasting hypoglycemia. It can combine with other autoimmune diseases, most commonly systemic lupus erythematosus. It usually occurs in women and accompanies acanthosis nigricans, hyperandrogenism, and, in many cases, ovary dysfunction. The diagnosis of type B insulin resistance syndrome is based largely on the presence of insulin receptor autoantibodies and hyperglycemia, or hypoglycemia and hyperinsulinemia. In some cases, patients with the type B insulin resistance have been successfully treated with immunosuppressive therapy and plasmapheresis. We experienced type B insulin resistance syndrome in a patient with chronic hepatitis B and used only plasmapheresis for treatment. The immunosuppressive therapy was omitted due to the state of activation of chronic hepatitis B. We present this case with a review of relevant literature.
- A Case of Ascites and Extensive Abdominal Distension Caused by Reversible Pulmonary Arterial Hypertension Associated with Graves' Disease.
-
Byoungho Choi, Young Sil Eom, Sei Hyun Kim, Hyun Seok Choi, Wook Jin Chung, Sihoon Lee
-
Endocrinol Metab. 2011;26(3):248-252. Published online September 1, 2011
-
DOI: https://doi.org/10.3803/EnM.2011.26.3.248
-
-
1,669
View
-
28
Download
-
1
Crossref
-
Abstract
PDF
- Patients with hyperthyroidism can develop left ventricular dysfunction and heart failure, but severe pulmonary hypertension association with hyperthyroidism is rarely seen. Herein, we describe the case of a 27-year-old female who presented with abdominal distension accompanied by pulmonary arterial hypertension and Graves' disease. Her pulmonary arterial hypertension was improved by treating the hyperthyroidism and pulmonary artery hypertension. Additionally, the patient's symptoms of right-side heart failure improved after pulmonary arterial pressure was reduced. Hyperthyroidism should be regarded as a reversible cause of associated pulmonary arterial hypertension.
-
Citations
Citations to this article as recorded by - A Case of Pulmonary Hypertension Recurred by Graves’ Disease
Jun Seop Lee, Young Sik Choi, Jae Woo Lee, Jin Seok Yoo, Youn Jung Choi, Dong Hyun Park
Kosin Medical Journal.2013; 28(2): 171. CrossRef
- Mutational Analysis of the NF1 Gene in Two Families with Neurofibromatosis 1 Accompanied by Pheochromocytoma.
-
Hyon Seung Yi, Sei Hyun Kim, Jihoon Kim, Eun Jin Bae, Suntaek Hong, Ie Byung Park, Yu Jin Kim, Sihoon Lee
-
Endocrinol Metab. 2011;26(2):177-184. Published online June 1, 2011
-
DOI: https://doi.org/10.3803/EnM.2011.26.2.177
-
-
1,897
View
-
25
Download
-
2
Crossref
-
Abstract
PDF
- Neurofibromatosis type 1 (NF1) is one of the most common autosomal dominant inherited disorders affecting the nervous system. NF1 is associated with mutations in the NF1 gene, which is located on chromosome sub-band 17q11.2 and contains 57 exons spanning approximately 300 kb of genomic DNA. NF1 is caused by a loss of function mutation of the NF1 gene, a tumor suppressor gene, which encodes for neurofibromin, a GTPase-activating protein (GAP) involved in the negative regulation of Ras activity. The GAP-related domain, which is encoded for by exons 20-27a, is one of the most important functional domains in neurofibromin. The cysteine-serine-rich domain has been recognized as an important functional domain in NF1-related pheochromocytomas. As the result of many genetic analyses of NF1-related pheochromocytomas, pheochromocytoma has generally been recognized as a true component of NF1. We recently experienced two families with NF1 accompanied by pheochromocytoma. The proband of family 1 is a 31-year-old female diagnosed with NF1 and pheochromocytoma. Gene analysis of the proband and her sister showed that the mutation of the NF1 gene (c.7907+1G>A) led to the skipping of exon 53 during NF1 mRNA splicing. The proband of family 2 is a 48-year-old male who was diagnosed with the same condition. Gene analysis demonstrated the mutation of the NF1 gene (c.5206-8C>G) with missplicing of exon 37. These novel germline mutations did not fall into the GAP-related nor the cysteine-serine-rich domains, but into the C-terminal area of the NF1 gene. This suggests that the correlation between the genotype and phenotype of NF1-related pheochromocytoma is somewhat difficult to characterize. Further studies will be necessary to confirm the function of the C-terminal area of the NF1 gene and its contribution to the development of NF1 and pheochromocytoma.
-
Citations
Citations to this article as recorded by - Mutation Spectrum of NF1 and Clinical Characteristics in 78 Korean Patients With Neurofibromatosis Type 1
Jung Min Ko, Young Bae Sohn, Seon Yong Jeong, Hyon-Ju Kim, Ludwine M. Messiaen
Pediatric Neurology.2013; 48(6): 447. CrossRef - Oncologic manifestations of neurofibromatosis type 1 in Korea
Eui Tae Kim, Hwan Namgung, Hyun Deok Shin, Soon Il Lee, Jee Eun Kwon, Myung Chul Chang, Dong Guk Park
Journal of the Korean Surgical Society.2012; 82(4): 205. CrossRef
- A Case of Primary Hyperparathyroidism Caused by Solitary Parathyroid Adenoma That was Not Detected by Both Ultrasonography and Sestamibi Scan.
-
Kyong Yong Oh, Byoungho Choi, Yukyung Lee, Do Hwan Kim, Hyon Seung Yi, Kwang Jun Kim, Sihoon Lee, Sung Kil Lim
-
Endocrinol Metab. 2011;26(2):166-170. Published online June 1, 2011
-
DOI: https://doi.org/10.3803/EnM.2011.26.2.166
-
-
1,804
View
-
24
Download
-
1
Crossref
-
Abstract
PDF
- Thanks to advances in assay techniques and routine measurements in serum chemical analysis, primary hyperparathyroidism has become far more frequently detected, and the number of asymptomatic patients has substantially increased. In the majority of patients (85%), a solitary adenoma is the underlying cause of primary hyperparathyroidism. Surgical excision is the treatment of choice for most cases of primary hyperparathyroidism; this procedure has a relatively high success rate. In the past decade, improvements in preoperative imaging have played a major role in a targeted operative approach, which allows for minimally invasive surgery to be performed. The success of parathyroid surgery depends on the accurate preoperative localization of parathyroid adenoma. In this study, we report the case of a 54 year-old woman with primary hyperparathyroidism who presented with left buttock and leg pain. For localization of the parathyroid lesion, an ultrasonography and a 99mTc-sestamibi scan were initially performed, but these attempts failed to localize the lesion. We then carried out contrast-enhanced CT; thereafter, a single parathyroid adenoma was detected. Therefore, in patients with negative results on both ultrasonography and 99mTc-sestamibi scan, contrast-enhanced CT may prove helpful for preoperative parathyroid localization.
-
Citations
Citations to this article as recorded by - Primary Hyperparathyroidism with Ectopic Parathyroid Adenoma Detected by Both99mTc-MIBI SPECT and Contrast-Enhanced Neck CT
Hye Jin Lim, Dong Geum Shin, Jun Bong Kim, Jin Taek Kim, Hyo Jeong Kim, Man Sil Park, Ho Jeong Lee
Korean Journal of Medicine.2012; 83(5): 641. CrossRef
- A Case of Aldosterone-secreting Adrenocortical Carcinoma.
-
Byoungho Choi, Sihoon Lee
-
Endocrinol Metab. 2011;26(1):36-37. Published online March 1, 2011
-
DOI: https://doi.org/10.3803/EnM.2011.26.1.36
-
-
Abstract
PDF
- No abstract available.
- Molecular Understanding and Assessment of Hypoparathyroidism.
-
Hyon Seung Yi, Byoungho Choi, Sihoon Lee
-
Endocrinol Metab. 2011;26(1):25-32. Published online March 1, 2011
-
DOI: https://doi.org/10.3803/EnM.2011.26.1.25
-
-
1,624
View
-
29
Download
-
1
Crossref
-
Abstract
PDF
- No abstract available.
-
Citations
Citations to this article as recorded by - Genetic and Clinical Characteristics of Korean Patients with Isolated Hypoparathyroidism: From the Korean Hypopara Registry Study
So Young Park, Young Sil Eom, Byoungho Choi, Hyon-Seung Yi, Seung-Hee Yu, Kiyoung Lee, Hyun-Seok Jin, Yoon-Sok Chung, Tae Sik Jung, Sihoon Lee
Journal of Korean Medical Science.2013; 28(10): 1489. CrossRef
- A Case of Adrenal Actinomycosis that Mimicked a Huge Adrenal Tumor.
-
Eui Joo Kim, Hyon Seung Yi, Inku Yo, Sanghui Park, Kyoung Min Kim, Yoon Soo Park, Sihoon Lee, Yeun Sun Kim, Ie Byung Park
-
Endocrinol Metab. 2010;25(2):147-151. Published online June 1, 2010
-
DOI: https://doi.org/10.3803/EnM.2010.25.2.147
-
-
1,813
View
-
22
Download
-
1
Crossref
-
Abstract
PDF
- The incidence of adrenal incidentalomas has increased because imaging studies are now being more frequently performed, including abdominal sonography, CT and MRI. Although there is only a consensus on the treatment of adrenal incidentalomas from the National Institute of Health (NIH) conference 2003, it is generally accepted that surgical resection is required if there's any possibility of malignancy or functionality of the adrenal tumor. Abdominopelvic actinomycosis is a rare chronic progressive suppurative disease that is caused by gram-positive bacteria of the genus actinomyces, which is part of the normal flora of the oral cavity and gastrointestinal tract, with low virulence. Herein, we report on a case of adrenal actinomycosis that imitated a huge adrenal tumor in a 39-year-old women, and the adrenal actinomycosis was confirmed histologically only after adrenalectomy. To the best of our knowledge, this is the first Korean case report on actinomycosis that occurred in the adrenal gland.
-
Citations
Citations to this article as recorded by - Masking and misleading: concomitant actinomycosis and B-cell lymphoma – a case report and review of literature
Jo Anne Lim, Peng Shyan Wong, Kar Nim Leong, Kar Loon Wong, Ting Soo Chow
Scottish Medical Journal.2018; 63(4): 125. CrossRef
- Acromegaly with Diabetes Insipidus after Pituitary Tumor Removal: Successful Pregnancy and Delivery.
-
Sei Hyun Kim, Joo Il Kim, Yae Min Park, In Sik Won, Kwen Chul Shin, Yunjeong Jo, Sihoon Lee, Yeun Sun Kim, Ki Young Lee, Ie Byung Park
-
J Korean Endocr Soc. 2010;25(1):56-60. Published online March 1, 2010
-
DOI: https://doi.org/10.3803/jkes.2010.25.1.56
-
-
Abstract
PDF
- A 33-year-old woman visited our hospital because of oligomenorrhea. Acromegaly was diagnosed based on elevated insulin like growth factor-I (IGF-I) and paradoxical growth hormone (GH) rise in oral glucose tolerance test. Pituitary macroadenoma was detected on magnetic resonance imaging (MRI). The pituitary tumor was removed. Still, diabetes insipidus developed. We prescribed desmopressin and bromocriptine. Two months post-surgery, IGF-I was decreased and a combined pituitary function test was normal, except for the follicle stimulating hormone response. Residual tumor was detected on MRI. The bromocriptine dose was increased and treatment with the long-acting somatostatin analogue octreotide long acting release (LAR) was begun. After the fifth round of octreotide LAR, IGF-I was normalized. After the seventh round of octreotide LAR, the patient became pregnant. Bromocriptine and octreotide LAR were stopped, and desmopressin was continued. Successful delivery occurred at week 38 of pregnancy. The patient was discharged without any complications. Acromegaly is a disease caused by chronic GH hypersecretion, generally related to a somatotroph adenoma. Amenorrhea and menstrual irregularities are common in acromegaly. Pregnancy rarely occurs because chronic anovulation usually exists. When gonadotroph axis was preserved, the possibility of pregnancy in a woman of child-bearing age with acromegaly should be considered.
- A Case of Hashimoto's Thyroiditis Accompanied by Autoimmune Hepatitis Diagnosed with Liver Biopsy.
-
Young Jun Lee, Ji Yoon Sung, Sei Hyun Kim, Hyon Seung Yi, Yun Soo Kim, Sihoon Lee, Ie Byung Park
-
J Korean Endocr Soc. 2009;24(4):287-292. Published online December 1, 2009
-
DOI: https://doi.org/10.3803/jkes.2009.24.4.287
-
-
1,891
View
-
23
Download
-
2
Crossref
-
Abstract
PDF
- Autoimmune thyroid diseases, including Hashimoto's thyroiditis (HT), are common organ-specific autoimmune disorders that often occur in conjunction with other autoimmune diseases. Autoimmune hepatitis (AIH) is a relatively rare disease of unknown etiology. In this condition, progressive destruction of the liver parenchyma occurs. Without proper treatment with immunosuppressive agents, such as prednisone and azathioprine, this condition leads to cirrhosis and liver failure. Timely detection and appropriate treatment of the AIH is prerequisite for the long-term survival of affected patients. We report here a case of HT accompanied by AIH confirmed by liver biopsy. On the basis of this case report, we suggest that, a sustained elevation of aminotransferases refractory to thyroid dysfunction correction should result in a liver biopsy to differentiate AIH from other forms of liver dysfunction or secondary to thyroid disorders. Treatment should commence promptly.
-
Citations
Citations to this article as recorded by - Autoimmune Hashimoto thyroiditis with concomitant autoimmune hepatitis
Nevena Manevska, Natasa Stojkovska, Ljubica Tasheva, Marija Jovanovski-Srceva, Tanja Makazlieva, Sinisha Stojanoski
Archives of Public Health.2022;[Epub] CrossRef - A Case of Demyelinating Peripheral Neuropathy Associated with Hashimoto`s Thyroiditis
Jung Hwan Park M.D., Sang Mo Hong M.D., Chang Bum Lee M.D., Yong Soo Park M.D., Dong Sun Kim M.D., Woong Hwan Choi M.D., You Hern Ahn M.D.
Journal of the Korean Geriatrics Society.2011; 15(4): 234. CrossRef
- Assessment of Insulin Resistance and Its Clinical Application.
-
Jihoon Kim, Lee Young Lee, Sihoon Lee
-
J Korean Endocr Soc. 2009;24(2):75-83. Published online June 1, 2009
-
DOI: https://doi.org/10.3803/jkes.2009.24.2.75
-
-
2,190
View
-
52
Download
-
1
Crossref
-
Abstract
PDF
- No abstract available.
-
Citations
Citations to this article as recorded by - Case Report of Impaired Fasting Glucose Improved with Korean Medicine Treatment and Dietetic Therapy
Eun-mi Kim, Ki-tae Kim
The Journal of Internal Korean Medicine.2021; 42(2): 175. CrossRef
- A Case of Thyroid Papillary Cancer Associated with Familial Adenomatous Polyposis.
-
Sung Jae Shin, Hyun Joo Lee, So Hun Kim, Wan Sub Shim, Sihoon Lee, Yoo Mee Kim, Yumie Rhee, Tae Il Kim, Bong Soo Cha, Hyun Chul Lee, Sung Kil Lim
-
J Korean Endocr Soc. 2004;19(2):209-216. Published online April 1, 2004
-
-
-
Abstract
PDF
- Familial adenomatous polyposis (FAP) is an autosomal dominant syndrome, typically characterized by multiple colorectal adenomas and increased incidence of colorectal carcinomas if left untreated. It is caused by germline mutations of the adenomatous polyposis coli (APC) gene, which has been mapped on chromosome 5q21, and is accompanied by various benign and malignant extracolonic manifestations. The prevalence of thyroid tumors developing in patients with FAP is about 1~2%, are associated with FAP and have certain characteristics; mean age of tumor diagnosis at less than 30 years of age, the pathology is the papillary histiotype in more than 90% of cases, including a so-called cribriform- morular pattern, and multifocality is a frequent feature. In a genetic analysis, thyroid cancer in FAP usually has a mutation in the 5-portion of exon 15 between 778 and 1309, on chromosome 5q21. Also, the ret/PTC (especially ret/PTC1 and ret/PTC3) and p53 genes are thought probably to be associated with thyroid cancer in FAP patients. A case of familial adenomatous polyposis, accompanied by thyroid papillary cancer, was experienced in a 29 year-old female. She had hundreds of adenomas throughout the entire colon and congenital hypertrophy of the retinal pigment epithelium (CHRPE). The pathological finding of thyroid cancer was revealed as a mixture of cribriform, trabecular and papillary patterns. In a genetic analysis, she and her brother had a germline mutation of the APC gene at codon 1309. In Korea, there has been no previous case of cribriform-morular pattern and familial genetic analysis in FAP associated with thyroid cancer. Therefore, this case is reported, with a review of the literature
|